Dive Brief:
- The U.S. Food and Drug Administration has published its warning letter to iRhythm Technologies, revealing inspectors accused the company of failing to seek clearance before making major changes to permitted uses for its Zio AT heart monitor.
- iRhythm disclosed the warning letter at the end of May but only provided a brief summary of the allegations. Now, the FDA has published the full, lightly redacted document, triggering a 4% slide in the company’s share price.
- William Blair analysts emerged from talks with iRhythm management believing that “customers are unlikely to change their usage of Zio AT over time, making this a resolvable issue.”
Dive Insight:
iRhythm provided a high-level look at the FDA’s accusations late last month, revealing the warning letter alleged “nonconformities to regulations for medical devices, including medical device reporting requirements, relating to the company’s Zio AT System and medical device quality system requirements.” The publication of the warning letter by the FDA fills in the gaps in the story.
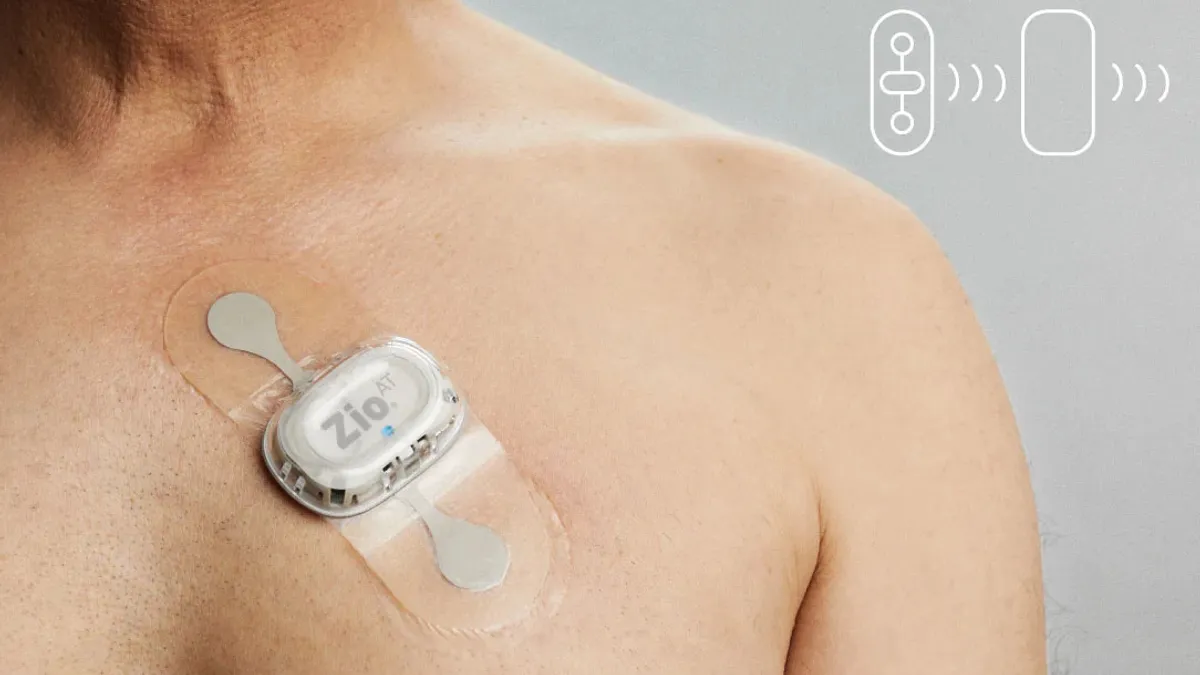
During an inspection of iRhythm’s Cypress, California, manufacturing facility in summer 2022, the FDA found iRhythm had changed the patient population for the Zio AT. The FDA cleared the device for long-term monitoring of arrhythmia events for non-critical care patients where real-time monitoring is not needed. According to the FDA, iRhythm began marketing Zio AT as a “mobile cardiac telemetry monitor” that can provide notifications “immediately” for the “near real-time monitoring” of “high-risk patients.”
“This change could significantly affect the safety or effectiveness of the device because it suggests that the device is intended for a new patient population – high-risk patients. High risk patients need near real-time monitoring because they are more likely to have a life-threatening arrhythmia, which requires timely treatment to prevent serious injury or death. Accordingly, these changes required the submission of a new 510(k),” the FDA wrote in the warning letter.
According to the FDA, iRhythm made changes to the hardware, firmware and algorithm that may require new 510(k) submissions. The FDA said changes to the algorithm “could significantly impact safety or effectiveness, such as through missed or incorrect detection of events or arrhythmias, which could lead to patient injury due to lack of treatment.” Similarly, the hardware and firmware changes “require new testing to support your assertion that the device’s safety and effectiveness is unaffected,” the FDA wrote.
The accusations echo the claims the FDA made against Abbott in a separate warning letter late last year. In that letter, the FDA accused Abbott of making “multiple significant design changes” to a heart disease test without filing 510(k) notifications about the revisions.
iRhythm is working to resolve the allegations, although the FDA deemed its initial responses to aspects including the targeting of high-risk patients to be inadequate. In a note to investors released after talking to iRhythm management, analysts at William Blair said the company thinks a “letter to file,” rather than a new 510(k) submission, is appropriate. If a full 510(k) filing is needed, iRhythm believes it could submit its paperwork within weeks, after which a 90-day review cycle would start.
Based on precedents, iRhythm believes it can keep the product on the market during the 510(k) process, according to the William Blair report. The analysts expect “time off the market would be limited to months” if iRhythm does need to withdraw the device. As such, the analysts think iRhythm can maintain share in a market targeted by Philips’ BioTelemetry, Boston Scientific’s Preventice Solutions and Baxter’s Bardy Diagnostics.
“Given our belief that the patient impact has been manageable and the company has been transparent and responsive, we believe that customers are unlikely to change their usage of Zio AT over time, making this a resolvable issue,” the analysts wrote. “Until we gain more information we will focus on catalysts. We continue to view the domestic opportunity as less than 35% penetrated, and we believe that the company can deliver on and exceed growth expectations.”
The rate at which the FDA has added Zio AT medical device reports to its database has increased over time, although that could simply reflect increased sales and more widespread use of the device. Of the 46 medical device reports for Zio AT in the FDA’s database, 31 were published since the start of last year. Eight were published before 2021, and the oldest report dates back to August 2018. iRhythm began selling Zio AT in 2017, initially through a “limited release.”