Dive Brief:
- Hologic received a warning letter in December from the Food and Drug Administration related to safety concerns with the company’s Biozorb implantable radiographic markers.
- The FDA found that Hologic failed to meet design requirements, including verifying the plastic spacer component of the markers is absorbed by the body. One complaint stated the device did not dissolve for almost five years, according to the warning letter, which the agency posted Tuesday.
- Hologic put a stop ship notice on the Biozorb device on Sept. 29, 2024, and will no longer manufacture the product, according to the warning letter. In October, Hologic notified customers that it would remove all unused markers from the market. A Hologic spokesperson wrote in an email that the company has been closely coordinating with the FDA and is “taking all necessary steps to resolve this matter in a comprehensive and timely manner.”
Dive Insight:
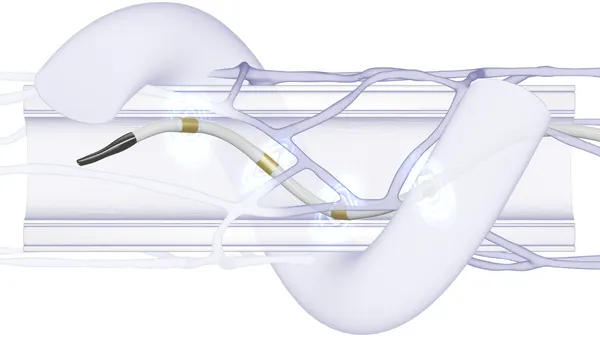
The Biozorb devices were implanted in breast tissue to mark tumor excision sites for monitoring or future treatments. They consist of a plastic component intended to dissolve into patients’ bodies and a permanent metal component.
Hologic acquired Biozorb in 2018 with its purchase of Focal Therapeutics. Since 2015, more than 91,500 of the devices have been sold, Hologic said in its latest letter to customers.
Biozorb was recalled in March and again in October of last year, due to adverse events including pain, infection, device migration and implant removal.
The warning letter follows an FDA inspection of Hologic’s Marlborough, Massachusetts, headquarters between July 30, 2024, and Sept. 24, 2024.
The FDA raised concerns that Biozorb’s device design did not address the intended patient population and anatomy types, as well as surgical requirements such as appropriate placement, fixation and depth. Design documents did not include a requirement on how radiation treatments can affect the implant’s performance and did not define how long it would take for the spacer material to be completely resorbed.
Hologic also lacked verification testing showing that the polylactic acid used in the spacer material is absorbed by the body, the FDA said.
The warning letter raised concerns with Hologic’s corrective and preventive action processes. Hologic received a “spike” of adverse events complaints in September 2023, including reports of breast pain, rashes, infection, scar tissue, tissue damage, device migration and explantation.
However, the company did not investigate the root cause of events that fell below the highest severity until May 2024, according to the warning letter.
The FDA listed cases of infection requiring antibiotic treatment or explantation that were reported after more than 30 days.
Hologic will conduct a retrospective review of all Biozorb complaints for reportability, the warning letter stated.
Although Biozorb is no longer sold, the FDA said it still has safety concerns for patients who have been implanted with the device. The agency said Hologic should identify patients who may be at risk for adverse events and benefit from having the implant removed.
Editor’s note: This article has been updated with comments from Hologic.